Análisis de clasificación no supervisada¶
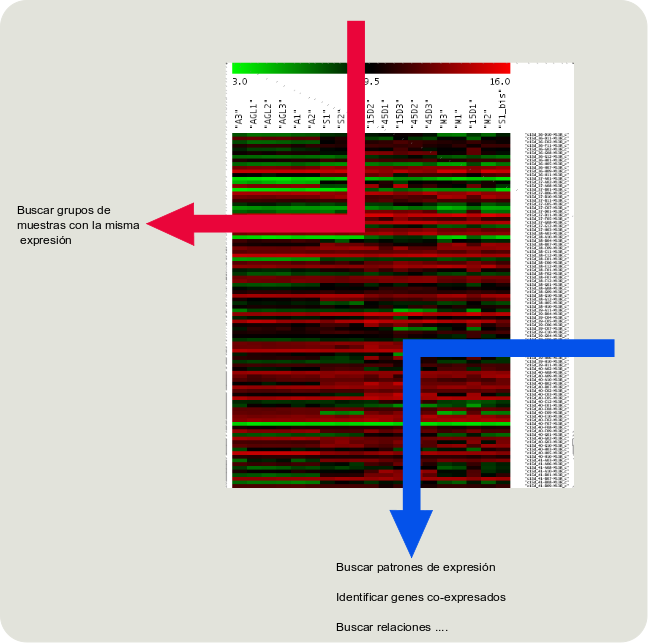
El clustering consiste en la clasificación de las muestras o de los genes según su nivel de similitud y nos permite encontrar relaciones desconocidas o verifican nuestras hipótesis. Lo podemos aplicar tanto a las muestras o a los genes.
Podemos clasificar las métodos de clustering en dos grupos:
Jerárquicos,nos indican una relación de dependencia entre los grupos: UPGMA, SOTA.
No jerárquicos, no nos indican una relación entre los grupos: K-MEANS, SOM, PCA.
Distancias¶
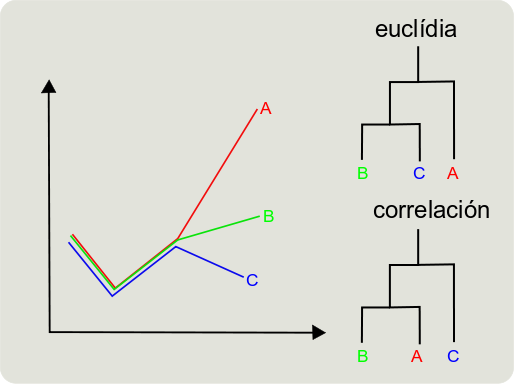
Pero todos ellos se basan en una medida de distancia para medir la similitud entre los grupos. Podemos utilizar varios tipos de distancias, euclídeas (absolutas) y tendencias (correlacionadas). Los grupos o clusters que encontremos dependerán de la distancia utilizada.
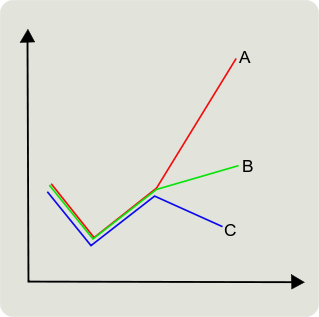
En distancia euclídea B es más cercana a C; pero en correlación B es más cercana a A.
La distancia euclídea es la distancia geométrica entre dos puntos, se representan los datos en el espacio multidimensional definido por las variables.

El coeficiente de correlación mide la linealidad entre dos valores de una muestra. Una de las más utilizadas es la correlación de Pearson.

Para evitar posibles efectos de las diferencias de expresión podemos normalizar respecto a la media de la expresión de cada gen.
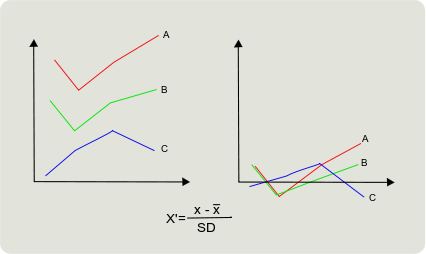
Dependiendo la distancia utilizada obtenemos diferentes clusters. La distancia euclídia nos sirve para ver relaciones entre las muestras viendo similitudes globales y la correlación para agrupar los genes según su patrón de expresión, ver las tendencias de la expresión de los genes. Tiene sentido que un gen se active o se inhiba más que otro a lo largo del desarrollo o respecto a varios tejidos. Pero no que un gen se activa o se inhiba entre dos muestras distintas, la expresión es más alta o más baja pero no hablamos de activación o inhibición.
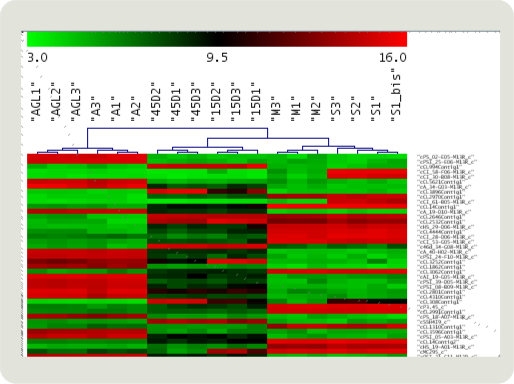
Agrupación de muestras¶
Utilizamos distancia euclídea (vemos la distancia global de una muestra respecto a otra y no tiene sentido ver si hay una tendencia de activación entre ellas).
Utilidades:
- Verificar triplicados (Nos deben salir juntos)
- Comprobar los grupos experimentales (Ej: muestras del mismo estadio)
- Detectar agrupaciones no conocidas (Ej: relaciones entre diferentes tejidos o estadios)
Problemas:
- Solo unos pocos genes varían el resto o son constantes o aleatorios. Serán cambios pequeños pero tendremos decenas de miles de ellos y nos pueden distorsionar el análisis.
- Con muestras al azar también haríamos un árbol. Necesitamos validar los resultados.
- La definición final de los clusters es subjetiva (¿Donde cortamos?)
Soluciones:
- Filtramos los genes constantes o con poca variación (diferenciales o por variabilidad del gen)
- Análisis de validación: Bootstrap , métodos específicos. Necesitamos una medida de la significación del cluster.
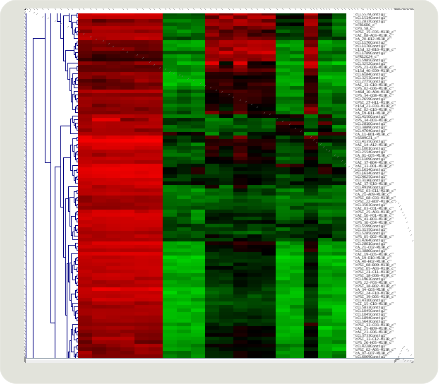
Agrupación de muestras¶
Utilizamos la correlación (Pearson), nos interesa ver si los cambios de expresión de los genes en diferentes condiciones, estadios, etc.
Utilidades:
- Detectar genes con el mismo patrón (¿corregulados?)
- Agrupar los distintos patrones para ver sus similitudes y diferencias
Problemas:
- Con muestras al azar también haríamos un árbol. Necesitamos validar los resultados.
- La definición final de los clusters es subjetiva (¿Donde cortamos?).
Programas de clustering¶
Podemos utilizar diferentes métodos de clustering métodos jerárquicos o particionados (no jerárquicos).
Un ejemplo de jerárquico es el UPGMA, en el los genes o las muestras se van a ordenar desde la pareja más cercana hasta la muestra más alejada produciendo un cladograma. Si gastamos UPGMA a partir de cada pareja calculara de nuevo el resto de distancias utilizando la media de la distancia esas dos entradas y así sucesivamente.
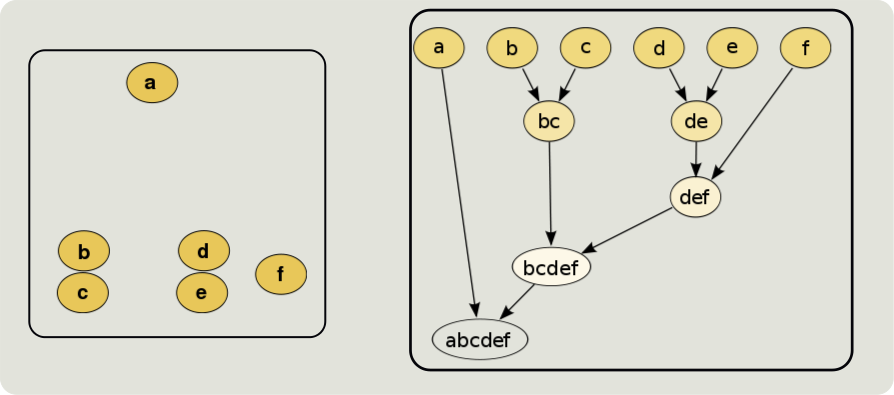
from http://en.wikipedia.org/wiki/Cluster_analysis#Hierarchical_clustering
Un ejemplo de particionado es el K-means, en el representamos los puntos en un espacio y determinamos a priori el número de cluster que vamos a utilizar. Entonces seleccionamos al azar tantas muestras como clusters queremos encontrar y asociamos a este las muestras más cercanas. Con está primera asociación se recalcula la media de las muestras de cada grupo y se sitúa en el espacio, a partir de este nuevo punto se reclasifican las muestras de cada cluster teniendo en cuenta la distancia al nuevo punto medio. Este proceso se vuelve a repetir hasta que los grupos se mantienen estables.
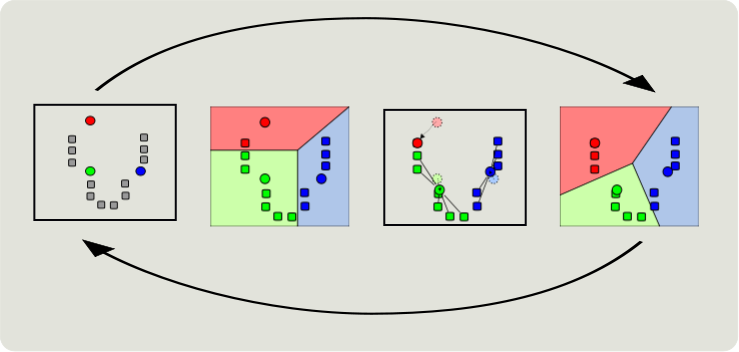
modified from http://en.wikipedia.org/wiki/Cluster_analysis#Hierarchical_clustering
Análisis de componentes principales¶
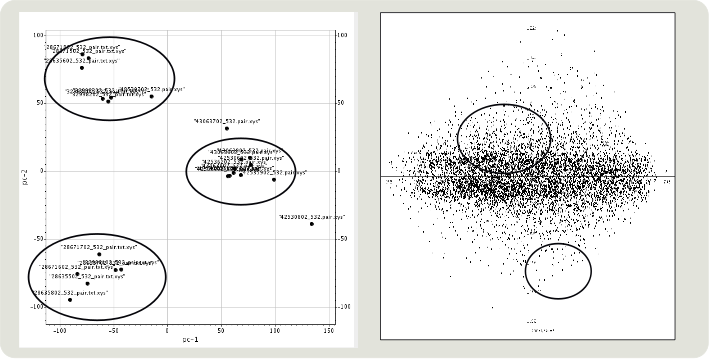
Otra forma de ver la relación entre las muestras o los genes es mediante el análisis de componentes principales o PCA. Este método no nos clasifica las muestras pero nos los muestra en un espacio de dos o 3 dimensiones en el cual nosotros podemos determinar la distancia y las relaciones entre ellos de forma muy intuitiva. Muy brevemente el método consiste en representar los datos en un espacio con tantas dimensiones como variables utilizadas y posteriormente buscar aquella proyección a planos de 2 dimensiones o espacios de 3 dimensiones que mejor expliquen la variabilidad existente.
El principal problema es encontrar los grupos ya que es un método subjetivo. Lo cual se complica cuando tenemos miles de puntos en el diagrama.