Análisis Funcionales¶
¿Y ahora qué hacemos con los datos?¶
El resultado de todos estos análisis es una serie de ficheros con datos como el nombre del gen, nivel de expresión, fold change, cluster, etc. Pero,¿Cómo analizamos estos datos?
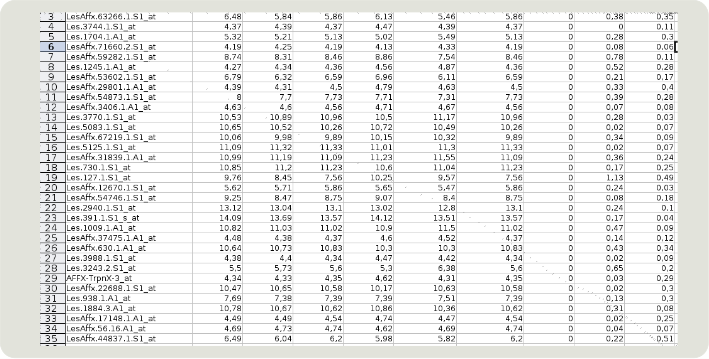
Tenemos información sobre la expresión de miles de genes en diferentes individuos, estadios o tejidos, es una cantidad enorme de información a la que hay que sacar partido. No tiene sentido hacer un experimento de microarray para fijarse únicamente en uno o unos pocos genes; para eso existen otras tecnologías más económicas y precisas. Para poder enfrentarse a esa cantidad de datos y poder conectarlos con los datos existentes se ha desarrollado las llamadas tecnologías de la información o más concretamente minería de datos (data mining).
Gracias a estas tecnologías podemos abordar los problemas de una forma global que nos permite acercarnos más a la realidad. El efecto de la expresión de un gen depende de muchos factores como: que variante de transcrito se expresa, el tiempo de vida media del mensajero, su traducción a proteína, la modificación de ésta, su interacción con otras moléculas, la variabilidad existente en este gen y en los relacionados, etc.
Las nuevas tecnologías de alto rendimiento y la minería de datos han modificado el proceso científico y hay que tenerlo en cuenta para evitar caer en asociaciones a posteriori.
El método científico se basa en la observación, realización de un hipótesis y en la validación o rechazo de la hipótesis mediante la experimentación.
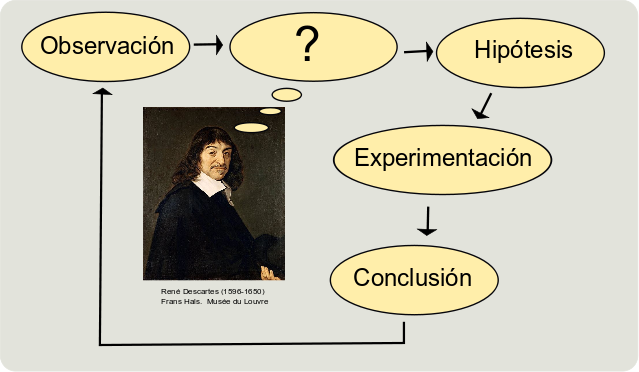
En cambio, cuando hacemos un experimento de microarrays, podemos observar el comportamiento de miles de genes y a partir de esos datos llegar a conclusiones; pero eso es erróneo hemos creado una idea una vez visto los datos y eso no es ciencia. Lo que debemos hacer es observar y analizar los datos y a partir de aquí establecer nuevas hipótesis que tendrán que ser validadas. Es muy fácil obtener asociaciones a posteriori. Eso no significa que los resultados no sean válidos, nos proporcionan nuevos datos y nuevas preguntas a responder.

Un caso distinto es utilizar un experimento de microarrays para probar una hipótesis previa. Por ejemplo: En antesis la división celular está ralentizada en el carpelo y se activará después de la polinización. Podemos analizar mediante microarray la expresión de los genes relacionados con ciclo celular y comprobar la hipótesis.
En resumen nos enfrentamos a varios problemas que debemos tener claro antes de comenzar los análisis funcionales:
- Tenemos un conjunto de datos y hay que analizarlo en su conjunto.
- No tiene mucho sentido hacer un array para fijarse luego en un sólo gen.
- Hay que tener en cuenta que es fácil que por azar encontremos asociaciones, hay que validar estadísticamente nuestros resultados. Las conclusiones que obtengamos en muchos casos tendrán que ser validadas por otros métodos, son en realidad hipótesis.
- Muchas veces los resultados obtenidos en los análisis funcionales no son tan espectaculares como desearíamos. Al menos confirman hipótesis si las teníamos o nos proporcionan información para realizar las hipótesis.
La Gene Ontology¶
Existen varias formas de estructurar el conocimiento y una de ellas son las ontologías. Nos vamos a centrar en la más utilizada para la descripción de la función: la Gene Ontology (GO).
Una ontología, en bioinformática, es un modo de estructurar el conocimiento.
La definición exacta en inglés es “Information Science: the hierarchical structuring of knowledge about things by subcategorizing them according to their essential (or at least relevant and/or cognitive) qualities”.
En las bases de datos hay una gran cantidad de información, pero si no se organiza de forma estructurada es difícil tratarla de modo sistemático.
Supongamos que queremos buscar todos los genes implicados en el metabolismo de lípidos, ¿qué buscamos? lipid, lipid metabolism, fat degradation, etc...
Este problema se puede solucionar mediante un vocabulario controlado. Existen varias ontologías relacionadas con la biología, pero la principal es la Gene Ontology (GO).
El proyecto pretende describir de un modo consistente los genes de distintas bases de datos.
Go se compone de tres ontologías dedicadas a los procesos biológicos, los componentes celulares y las funciones moleculares.
- Molecular function: la función que cumple el producto de un gen. Ejemplos: transcription factor, DNA helicase.
- Biological process: procesos, como “mitosis” o “metabolismo de purinas”.
- Cellular component: estructuras subcelulares, localizaciones, complejos macromoleculares. Ejemplos: núcleo, telómero, origin recognition complex
Un gen puede tener varios términos GO asociados en cada ontología. Los términos GO facilitan las búsquedas en las bases de datos y las búsquedas sistemáticas.
Los genes se anotan a un nivel u otro dependiendo de lo que se sepa sobre ellos.
Una ontología se parece a un diccionario, pero se diferencia en algunos aspectos.
Los términos están definidos de forma exacta y no hay sinónimos.
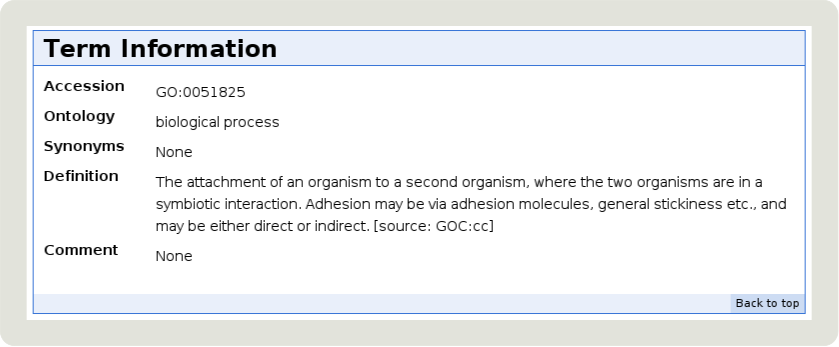
Los términos están ordenados en una estructura jerárquica.

La estructura de la ontología no es un árbol, se parece más a una red ya que hay conexiones laterales.
Análisis funcionales¶
El análisis consiste en comprobar si un grupo de genes (cluster, diferenciales...) hay algún término enriquecido respecto al conjunto de genes.
Necesitamos tener una anotación de los genes del microarray respecto a los términos GO. Pero luego hay que distinguir entre desviaciones de los porcentajes debidas al azar de las desviaciones significativas. Para ello hay que tener en cuenta los tamaños de cada grupo, el porcentaje existente de cada termino GO en los diferentes grupos, etc.
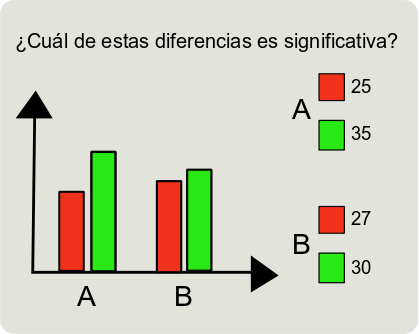
Hay diferentes programas y estadísticos que podemos utilizar. Hay que tener en cuenta que a parte del p-value nos dé un valor ajustado (FDR, Boniferri, etc).
- Fatigo
Programa de la suite Babelomics. Utilizando el test de Fisher, nos calcula un p-value ajustado con FDR. Puede utilizar anotaciones de su base de datos o nuestras propias anotaciones.
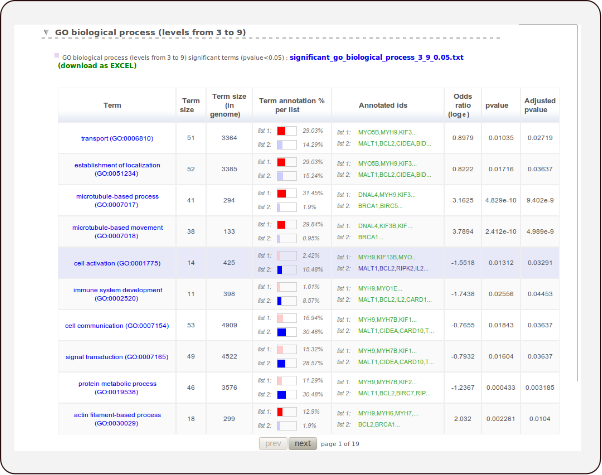
- FatiScan
Este programa de la suite Babelomics está diseñado para localizar funciones aunque no todos los genes estén sobre-expresados.Para tener una ruta activada no es necesario que se expresen más todos los genes de la ruta, basta con que se expresen.Partimos de una lista de genes ordenada respecto a un criterio biológico, por ejemplo fold-change.
imagen faiscan
- Blast2GO
El Blast2GO es un programa diseñado para crear anotaciones a partir de datos de secuencia; pero también contiene diferentes análisis. Entre ellos un test de Fisher. Hace el análisis usando la profundidad de la jerarquía de los términos GO. Las anotaciones contienen el nivel más bajo en el que este anotado el gen. Este programa tiene en cuenta la jerarquía y nos muestra los términos agrupados más concretos que sean diferenciales.
imagen blast2go y datos<jerarquia
Limitaciones de los análisis funcionales¶
Muchas veces los análisis muestras muy pocos resultados significativos, sobretodo si usamos correctamente los correctores del p-value, o solo términos muy generales. Esto suele estar asociado a dos tipos de problemas, el nivel de anotación de nuestro microarray y al número de genes que estemos analizando. Por ello, sobretodo si trabajamos en especies con anotaciones parciales, tendremos que recurrir muchas veces a la búsqueda manual de estas funciones diferenciadas. Pero hay que tener siempre en cuenta que en ese caso no tenemos apoyo estadístico y que , mas que nunca, se tratará de hipótesis que habrá que contrastar.