3 Lectura y normalización¶
Captación de la imagen¶
Lo que van a proporcionar los equipos una vez hibridados y detectados los microarrays es una imagen de intensidad de luz.

La captación de la imagen depende del sistema utilizado y normalmente se realiza por el servicio de hibridación. Lo que va a realizar es:
- Comprobar la hibridación
- Identificar los puntos
- Cuantificar la intensidad del punto
- Cuantificar el fondo del array
- Dar un valor de intensidad media para cada punto.
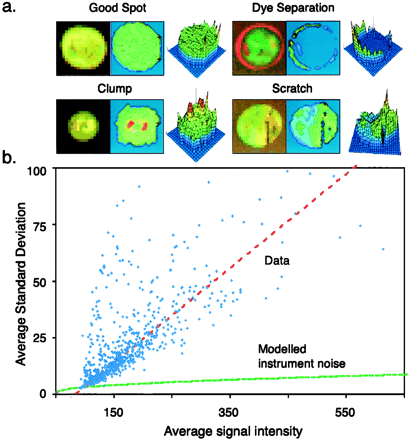
from C. Brown et al. Image metrics in the statistical analysis of DNA microarray data. PNAS July 31, 2001 vol.98 no. 16 http://www.pnas.org/content/98/16/8944.full
El resultado de la captación es un fichero de texto con las coordenadas de la posición del punto y su intensidad.

Pero hay variaciones experimentales que afectan a estas lecturas, como por ejemplo: fondo en las hibridaciones, manchas o diferentes intensidades entre microarrays.
Por lo que es necesario solucionar estos problemas. Eliminar el ruido de fondo y poner tanto las distintas zonas del microarray como los distintos microarrays a la misma escala y punto cero. A este proceso se denomina normalización y depende del sistema utilizado.
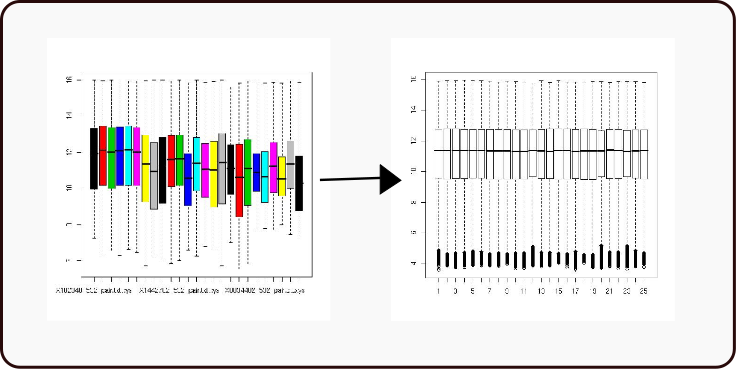
Normalización¶
Este proceso puede ser realizado tanto por el servicio de hibridación como por el usuario y va a permitir la comparación de intensidades entre las distintas sondas del microarrays y entre microarrays. La normalización se puede basar en distintos datos:
- Intensidad del punto
- Intensidad media del array
- Puntos control
- Genes “House keeping”
- Distribuciones de intensidad
- Se supone que la mayoría de genes se van a expresar igual en las diferentes muestras y que habrán tanto sobre-expresados como reprimidos
Existen diferentes métodos para los diferentes sistemas de microarrays. Como por ejemplo:
- Lowess para arrays de dos colores
Esperamos que si hibridamos la misma sonda marcada con los esperaríamos que: [Rojo]=[Verde].
[Rojo]/[Verde]=2 <—–> [Verde]/[Rojo]=0.5
o usando log2
log2([Rojo]/[Verde])=1 <—–> log2( [Verde]/[Rojo])=-1
Pero eso no sucede debido a los diferentes propiedades del marcaje. La normalización de Lowess tiene en cuenta esto y que la variación cambia según la intensidad. Hace regresiones en cada región del array y el modelo se basa en el log(Rojo/Verde).
M = log(R) - log(V)= log(R/V)
A = (log(R) + log(V))/2
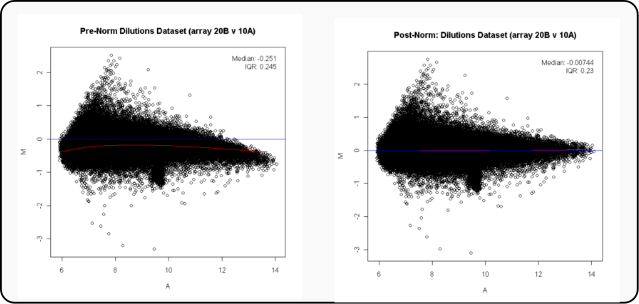
from: http://en.wikipedia.org/wiki/MA_plot
- MAS5 (Affymetrix)
Se basa en las sondas MM y PM
Se basa en la comparación de las sondas PM (perfect match) y las MM (mismatched match) que presentan los microarrays de Affymetrix.
- GAACTGAGACATGATA GAACTGACACATGATA
Con este sistema de dos sondas se intenta paliar la hibridación cruzada entre las diferentes sondas. Luego se selecciona el array más cercano a la media y lo utiliza como referencia, ajustando los niveles medios de cada array al de ese array
- RMA (Affymetrix y oligonucelótidos)
Se basa únicamente en las sondas PM (perfect match). Las sondas MM mezclan fondo y señal de hibridación, añaden variación y efectos de secuencia específicos. El RMA ajusta las intensidades del fondo (ajusta el nivel cero de todos lo arrays). Normaliza mediante quantiles las señales PM ajustadas.(hace que todos los arrays tengan la misma distribución de intensidades). Transforma la intensidad en logaritmos de base 2.

El resultado de la normalización es un fichero de texto con el valor de la expresión media, en escala logarítmica en base 2, de cada gen en las diferentes muestras. Se habrán eliminado los controles de fondo y se nos da la media de intensidad de las diferentes sondas de cada transcrito. Pero las herramientas utilizadas durante este proceso también nos sirven detectar aquellas hibridaciones no válidas; por ejmplo que tengan grandes manchas, muy poca señal, etc. Estas hibridaciones tienen que ser eliminadas, ya que nos distorsionan la normalización.